# Imports
import pymc3 as pm
import numpy.random as npr
import numpy as np
import matplotlib.pyplot as plt
import matplotlib as mpl
from collections import Counter
import seaborn as sns
import missingno as msno
# Set plotting style
# plt.style.use('fivethirtyeight')
sns.set_style('white')
sns.set_context('poster')
%load_ext autoreload
%autoreload 2
%matplotlib inline
%config InlineBackend.figure_format = 'retina'
import warnings
warnings.filterwarnings('ignore')
import pyqrcode as pq
icon = pq.create('https://ericmjl.github.io/bayesian-stats-talk')
icon.png('images/qrcode.png', scale=6)
Bayesian Statistical Analysis with PyMC3¶
Eric J. Ma, MIT Biological Engineering, Insight Health Data Science Fellow, NIBR Data Science
PyCon 2017, Portland, OR
- HTML Notebook on GitHub: ericmjl.github.io/bayesian-stats-talk
- Twitter: @ericmjl
talk features¶
- minimal field jargon: let's focus on the mechanics of analysis, rather than the terminology. e.g. won't explain A/B testing, spike & slab regression, conjugate distributions...
- pareto principle: the basics will get you to 80% of what you'll need
- enjoy the talk: focus on Bayes, get code later!
assumed knowledge¶
- familiarity with Python:
- objects & methods
- context manager syntax
- knowledge of basic stats terminology:
- mean
- variance
- interval
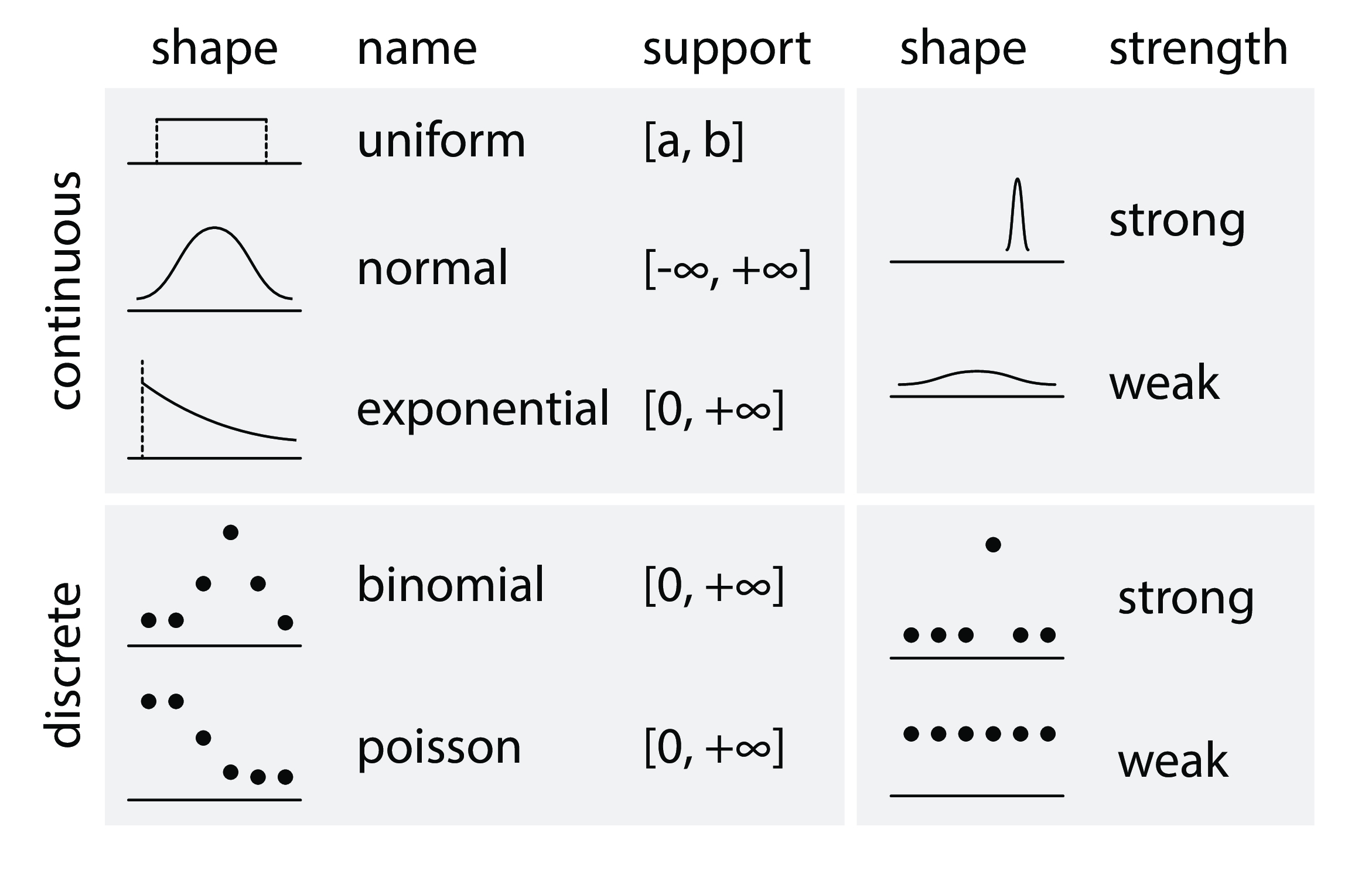
the obligatory Bayes rule slide¶
- $ P(H|D) $: Probability that the hypothesis is true given the data.
- $ P(D|H) $: Probability of the data arising given the hypothesis.
- $ P(H) $: Probability that the hypothesis is true, globally.
- $ P(D) $: Probability of the data arising, globally.
bayesian thinking¶
update beliefs having seen the evidence
pymc3¶
![]()
- Library of statistical distributions, sampling algorithms, and syntax for specifying statistical models
- Everything in Python!
computation-powered Bayesian stats¶
- Bayesian statistics was infeasible because of complicated integrals needed to compute posterior distributions.
- Markov Chain Monte Carlo (MCMC) sampling enables us to estimate shape of posterior distributions; calculus not required.
common statistical analysis problems¶
- parameter estimation: "is the true value equal to X?"
- comparison between experimental groups: "are the treatments different from the control(s)?"
problem type 1: parameter estimation¶
"is the true value equal to X?"
OR
"given the data, for the parameter of interest, what is the probability distribution over the possible values?"
example 1: the obligatory coin toss problem¶
I tossed my coin $ n $ times, and it came up as heads $ h $ times. Is it biased?
parameterized problem¶
"I want to know $ p $, the probability of tossing heads. Given $ n $ tosses and $ h $ observed heads, is it probable that the value of $ p $ is close to $ 0.5$ , say, in the interval $[0.48, 0.52]$?"
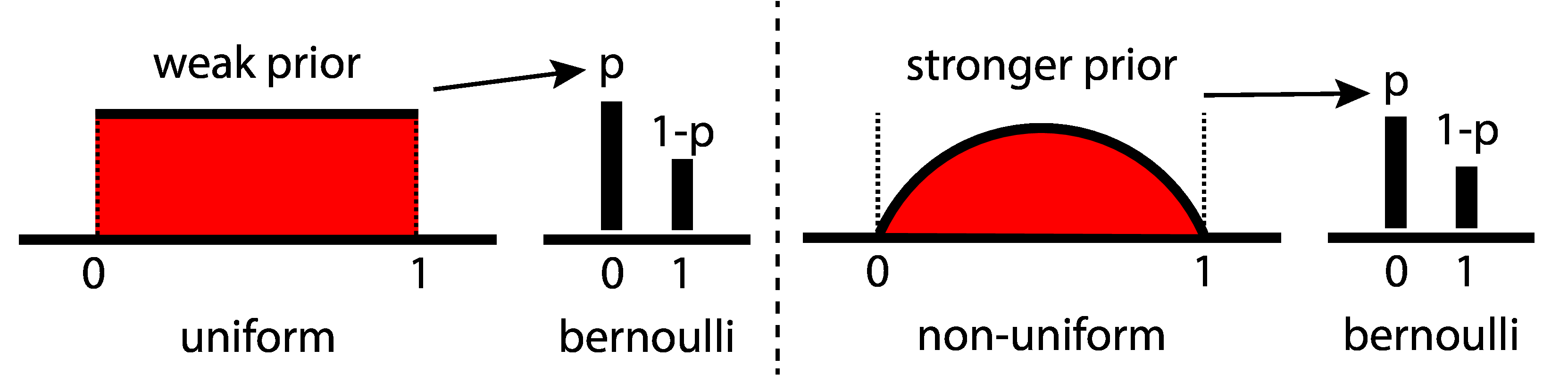
prior¶
- prior belief about parameter: $ p \sim Uniform(0, 1) $
- likelihood function: $ data \sim Bernoulli(p) $
# Make the data needed for the problem.
from random import shuffle
total = 30
n_heads = 11
n_tails = total - n_heads
tosses = [1] * n_heads + [0] * n_tails
shuffle(tosses)
data¶
print(tosses)
def plot_coins():
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
ax.bar(list(Counter(tosses).keys()), list(Counter(tosses).values()))
ax.set_xticks([0, 1])
ax.set_xticklabels(['tails', 'heads'])
ax.set_ylim(0, 20)
ax.set_yticks(np.arange(0, 21, 5))
return fig
fig = plot_coins()
plt.show()

code¶
# Context manager syntax. `coin_model` is **just**
# a placeholder
with pm.Model() as coin_model:
# Distributions are PyMC3 objects.
# Specify prior using Uniform object.
p_prior = pm.Uniform('p', 0, 1)
# Specify likelihood using Bernoulli object.
like = pm.Bernoulli('likelihood', p=p_prior,
observed=tosses)
# "observed=data" is key
# for likelihood.
MCMC Inference Button (TM)¶
with coin_model:
# don't worry about this:
step = pm.Metropolis()
# focus on this, the Inference Button:
coin_trace = pm.sample(2000, step=step)
results¶
pm.traceplot(coin_trace)
plt.show()

pm.plot_posterior(coin_trace[100:], color='#87ceeb',
rope=[0.48, 0.52], point_estimate='mean',
ref_val=0.5)
plt.show()

- 95% highest posterior density (HPD) encompasses the region of practical equivalence (ROPE).
- GET MORE DATA!
pattern¶
- parameterize your problem using statistical distributions
- justify your model structure
- write model in PyMC3, hit the Inference ButtonTM
- interpret based on posterior distributions
- (optional) with new information, modify model structure.
example 2: the chemical activity problem¶
I have a newly developed molecule X; how good is X in stopping flu replication?
experiment¶
test a range of concentrations of X, measure flu activity
compute IC50: the concentration of X that causes the replication rate of the virus to be halved.
data¶
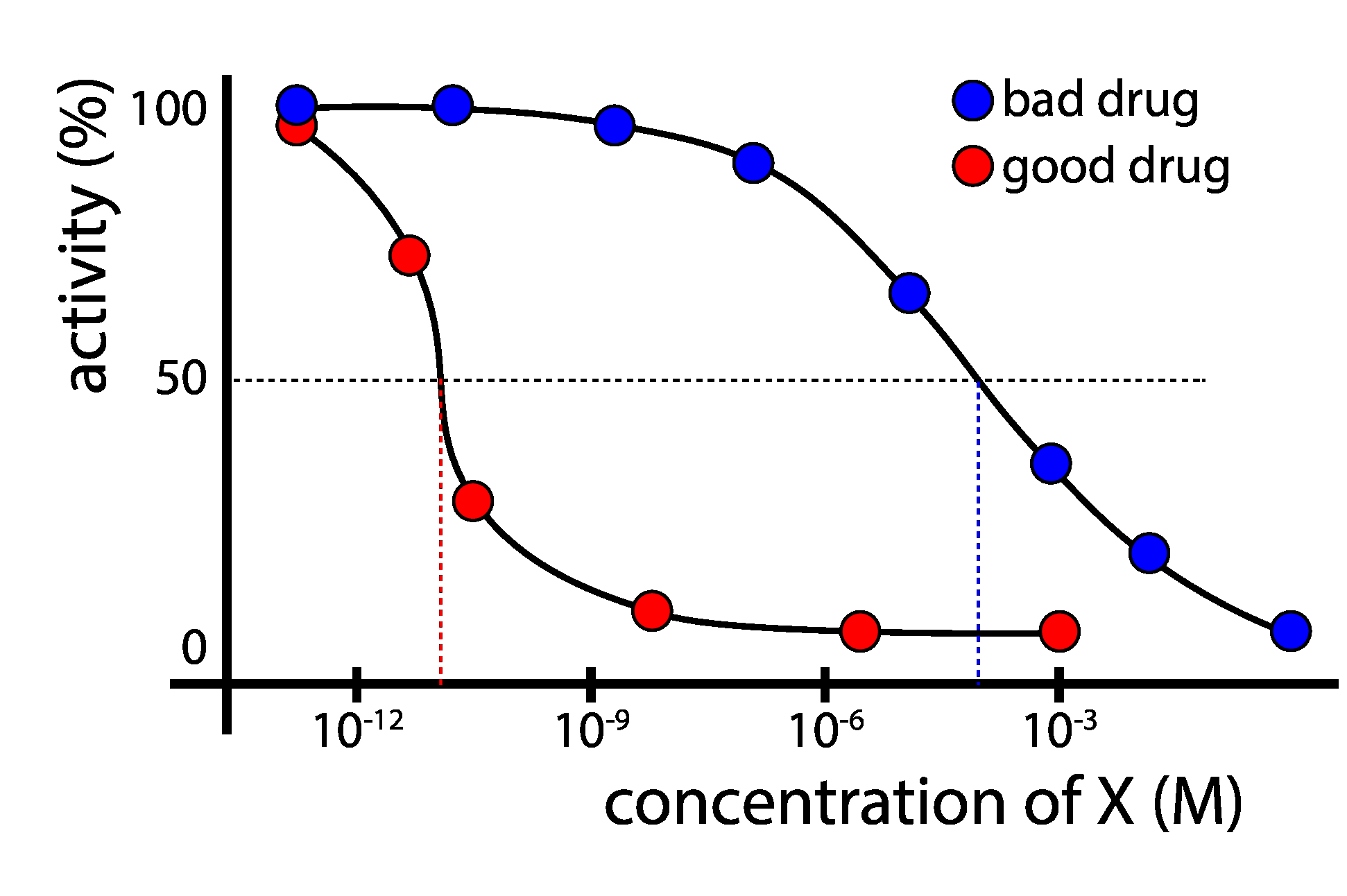
import numpy as np
chem_data = [(0.00080, 99),
(0.00800, 91),
(0.08000, 89),
(0.40000, 89),
(0.80000, 79),
(1.60000, 61),
(4.00000, 39),
(8.00000, 25),
(80.00000, 4)]
import pandas as pd
chem_df = pd.DataFrame(chem_data)
chem_df.columns = ['concentration', 'activity']
chem_df['concentration_log'] = chem_df['concentration'].apply(lambda x:np.log10(x))
# df.set_index('concentration', inplace=True)
parameterized problem¶
given the data, what is the IC50 value of the chemical, and the uncertainty surrounding it?
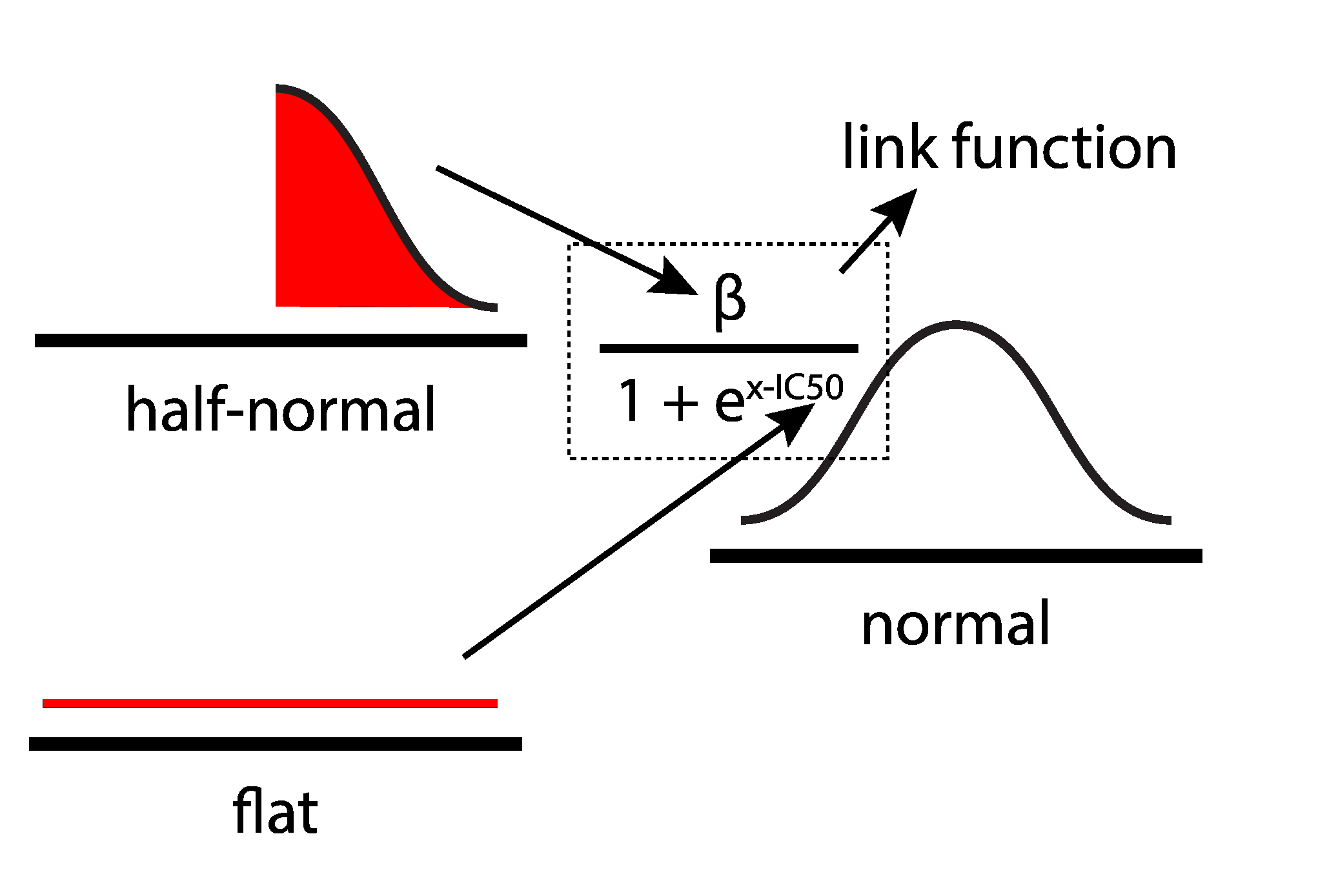
prior¶
- measurement function from domain knowledge: $ m = \frac{\beta}{1 + e^{x - IC_{50}}} $
- prior belief about constant to be estimated: $ \beta \sim HalfNormal(100^2) $
- prior belief about parameter of interest: $ log(IC_{50}) \sim ImproperFlat $
- likelihood function: $ data \sim N(m, 1) $
data¶
def plot_chemical_data(log=True):
fig = plt.figure(figsize=(10,6))
ax = fig.add_subplot(1,1,1)
if log:
ax.scatter(x=chem_df['concentration_log'], y=chem_df['activity'])
ax.set_xlabel('log10(concentration (mM))', fontsize=20)
else:
ax.scatter(x=chem_df['concentration'], y=chem_df['activity'])
ax.set_xlabel('concentration (mM)', fontsize=20)
ax.set_xticklabels([int(i) for i in ax.get_xticks()], fontsize=18)
ax.set_yticklabels([int(i) for i in ax.get_yticks()], fontsize=18)
plt.hlines(y=50, xmin=min(ax.get_xlim()), xmax=max(ax.get_xlim()), linestyles='--',)
return fig
fig = plot_chemical_data(log=True)
plt.show()

code¶
with pm.Model() as ic50_model:
beta = pm.HalfNormal('beta', sd=100**2)
ic50_log10 = pm.Flat('IC50_log10') # Flat prior
# MATH WITH DISTRIBUTION OBJECTS!
measurements = beta / (1 + np.exp(chem_df['concentration_log'].values -
ic50_log10))
y_like = pm.Normal('y_like', mu=measurements,
observed=chem_df['activity'])
# Deterministic transformations.
ic50 = pm.Deterministic('IC50', np.power(10, ic50_log10))
MCMC Inference Button (TM)¶
with ic50_model:
step = pm.Metropolis()
ic50_trace = pm.sample(100000, step=step)
pm.traceplot(ic50_trace[2000:], varnames=['IC50_log10', 'IC50']) # live: sample from step 2000 onwards.
plt.show()

results¶
pm.plot_posterior(ic50_trace[4000:], varnames=['IC50'],
color='#87ceeb', point_estimate='mean')
plt.show()

The IC50 of the chemical is between approx. [2 mM, 2.4 mM] (95% HPD). It's kind of a bad chemical; uncertainty doesn't matter much here, because of the scale of badness...
problem type 2: comparison between treatment groups¶
"are my experimental treatments different from my controls?"
example 1: the drug IQ problem¶
does a drug treatment affect IQ scores?
(documented in Kruschke, 2013, example modified from PyMC3 documentation)
drug = [ 99., 110., 107., 104., 103., 105., 105., 110., 99.,
109., 100., 102., 104., 104., 100., 104., 101., 104.,
101., 100., 109., 104., 105., 112., 97., 106., 103.,
101., 101., 104., 96., 102., 101., 100., 92., 108.,
97., 106., 96., 90., 109., 108., 105., 104., 110.,
92., 100.]
placebo = [ 95., 105., 103., 99., 104., 98., 103., 104., 102.,
91., 97., 101., 100., 113., 98., 102., 100., 105.,
97., 94., 104., 92., 98., 105., 106., 101., 106.,
105., 101., 105., 102., 95., 91., 99., 96., 102.,
94., 93., 99., 99., 113., 96.]
def ECDF(data):
x = np.sort(data)
y = np.cumsum(x) / np.sum(x)
return x, y
def plot_drug():
fig = plt.figure()
ax = fig.add_subplot(1,1,1)
x_drug, y_drug = ECDF(drug)
ax.plot(x_drug, y_drug, label='drug, n={0}'.format(len(drug)))
x_placebo, y_placebo = ECDF(placebo)
ax.plot(x_placebo, y_placebo, label='placebo, n={0}'.format(len(placebo)))
ax.legend()
ax.set_xlabel('IQ Score')
ax.set_ylabel('Cumulative Frequency')
ax.hlines(0.5, ax.get_xlim()[0], ax.get_xlim()[1], linestyle='--')
return fig
# For my own curiosity: from a frequentist point of view, is there a "statistically significant" difference
# between the two treatments?
from scipy.stats import ttest_ind
ttest_ind(drug, placebo)
experiment¶
- randomly assign participants to two treatment groups:
+drugvs.-drug
- measure IQ score for each participant
priors¶
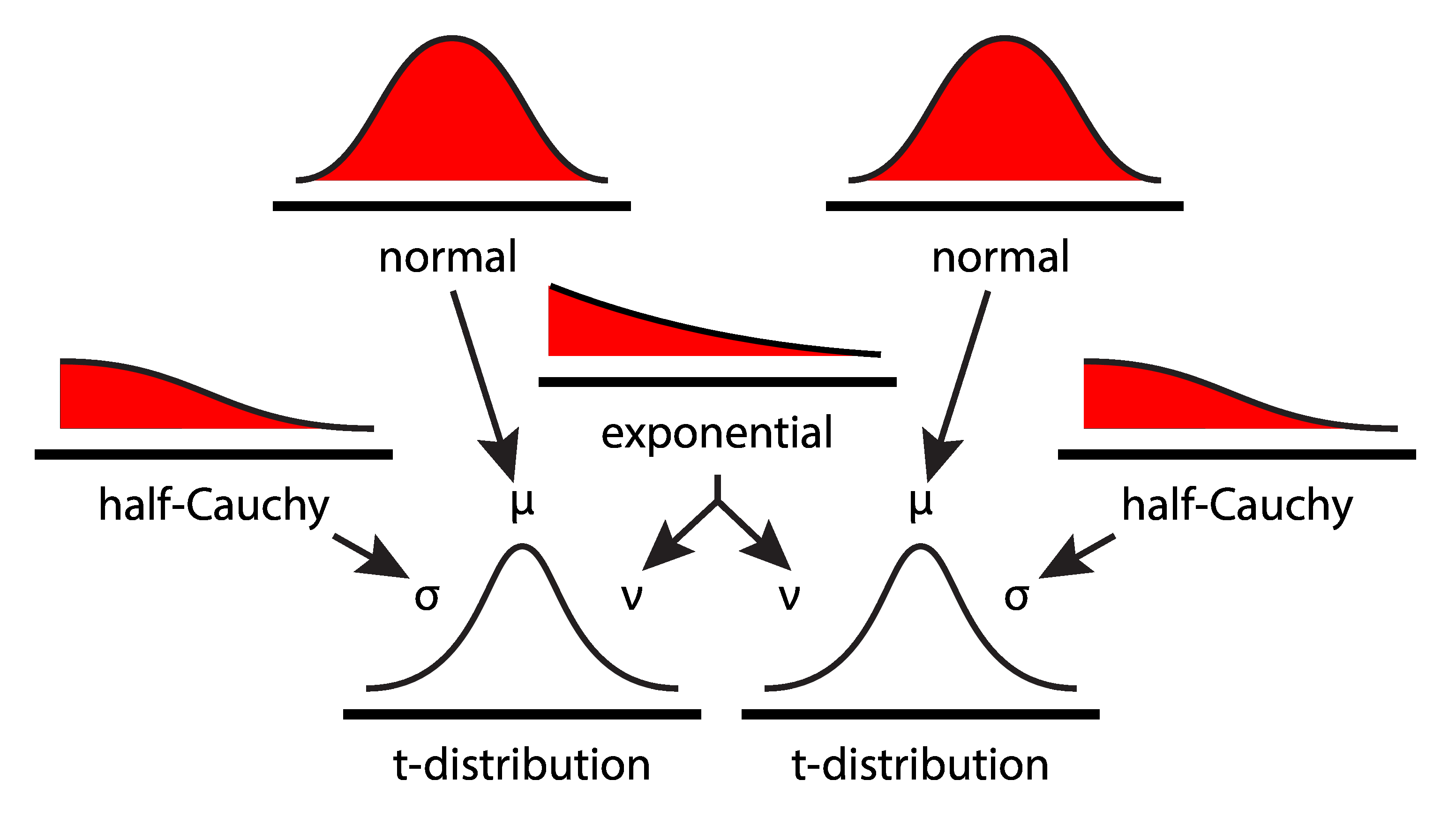
- measured data are t-distributed: $ data \sim StudentsT(\mu, \sigma, \nu) $
- means are normally distributed: $ \mu \sim N(0, 100^2) $
- degrees of freedom are exponentially-distributed: $ \nu \sim Exp(30) $
- variance is positively-distributed: $ \sigma \sim HalfCauchy(100^2) $
data¶
fig = plot_drug()
plt.show()

code¶
y_vals = np.concatenate([drug, placebo])
labels = ['drug'] * len(drug) + ['placebo'] * len(placebo)
data = pd.DataFrame([y_vals, labels]).T
data.columns = ['IQ', 'treatment']
with pm.Model() as kruschke_model:
# Focus on the use of Distribution Objects.
# Linking Distribution Objects together is done by
# passing objects into other objects' parameters.
mu_drug = pm.Normal('mu_drug', mu=0, sd=100**2)
mu_placebo = pm.Normal('mu_placebo', mu=0, sd=100**2)
sigma_drug = pm.HalfCauchy('sigma_drug', beta=100)
sigma_placebo = pm.HalfCauchy('sigma_placebo', beta=100)
nu = pm.Exponential('nu', lam=1/29) + 1
drug_like = pm.StudentT('drug', nu=nu, mu=mu_drug,
sd=sigma_drug, observed=drug)
placebo_like = pm.StudentT('placebo', nu=nu, mu=mu_placebo,
sd=sigma_placebo, observed=placebo)
diff_means = pm.Deterministic('diff_means', mu_drug - mu_placebo)
pooled_sd = pm.Deterministic('pooled_sd',
np.sqrt(np.power(sigma_drug, 2) +
np.power(sigma_placebo, 2) / 2))
effect_size = pm.Deterministic('effect_size',
diff_means / pooled_sd)
MCMC Inference Button (TM)¶
with kruschke_model:
kruschke_trace = pm.sample(10000, step=pm.Metropolis())
results¶
pm.traceplot(kruschke_trace[2000:],
varnames=['mu_drug', 'mu_placebo'])
plt.show()

pm.plot_posterior(kruschke_trace[2000:], color='#87ceeb',
varnames=['mu_drug', 'mu_placebo', 'diff_means'])
plt.show()

- Difference in mean IQ: [0.5, 4.6]
- Frequentist p-value: $ 0.02 $ (!!!!!!!!)
def get_forestplot_line(ax, kind):
widths = {'median': 2.8, 'iqr': 2.0, 'hpd': 1.0}
assert kind in widths.keys(), f'line kind must be one of {widths.keys()}'
lines = []
for child in ax.get_children():
if isinstance(child, mpl.lines.Line2D) and np.allclose(child.get_lw(), widths[kind]):
lines.append(child)
return lines
def adjust_forestplot_for_slides(ax):
for line in get_forestplot_line(ax, kind='median'):
line.set_markersize(10)
for line in get_forestplot_line(ax, kind='iqr'):
line.set_linewidth(5)
for line in get_forestplot_line(ax, kind='hpd'):
line.set_linewidth(3)
return ax
pm.forestplot(kruschke_trace[2000:],
varnames=['mu_drug', 'mu_placebo'])
ax = plt.gca()
ax = adjust_forestplot_for_slides(ax)
plt.show()

Forest plot: 95% HPD (thin line), IQR (thicker line), and median (dot) of the posterior distribution on the same axes, allowing us to directly compare the treatment and control.
def overlay_effect_size(ax):
height = ax.get_ylim()[1] * 0.5
ax.hlines(height, 0, 0.2, 'red', lw=5)
ax.hlines(height, 0.2, 0.8, 'blue', lw=5)
ax.hlines(height, 0.8, ax.get_xlim()[1], 'green', lw=5)
ax = pm.plot_posterior(kruschke_trace[2000:],
varnames=['effect_size'],
color='#87ceeb')
overlay_effect_size(ax)

- Effect size (Cohen's d, none to small, medium, large) could be anywhere from essentially nothing to large (95% HPD [0.0, 0.77]).
- Improvement in IQ is 0-4 points.
- The drug is very likely inconsequential.
- No evidence of biological significance.
example 2: the phone sterilization problem¶
compared to two commonly-used treatments, do my "fancy methods" sterilize my phone better?
the experiment design¶
- randomly assign phones to one of six groups: 4 "fancy" methods + 2 "control" methods.
- swab phone before and after treatment, grow bacteria
- count number of bacteria colonies formed, compare counts before and after
renamed_treatments = dict()
renamed_treatments['FBM_2'] = 'FM1'
renamed_treatments['bleachwipe'] = 'CTRL1'
renamed_treatments['ethanol'] = 'CTRL2'
renamed_treatments['kimwipe'] = 'FM2'
renamed_treatments['phonesoap'] = 'FM3'
renamed_treatments['quatricide'] = 'FM4'
# Reload the data one more time.
data = pd.read_csv('datasets/smartphone_sanitization_manuscript.csv', na_values=['#DIV/0!'])
del data['perc_reduction colonies']
# Exclude cellblaster data
data = data[data['treatment'] != 'CB30']
data = data[data['treatment'] != 'cellblaster']
# Rename treatments
data['treatment'] = data['treatment'].apply(lambda x: renamed_treatments[x])
# Sort the data according to the treatments.
treatment_order = ['FM1', 'FM2', 'FM3', 'FM4', 'CTRL1', 'CTRL2']
data['treatment'] = data['treatment'].astype('category')
data['treatment'].cat.set_categories(treatment_order, inplace=True)
data['treatment'] = data['treatment'].cat.codes.astype('int32')
data = data.sort_values(['treatment']).reset_index(drop=True)
data['site'] = data['site'].astype('category').cat.codes.astype('int32')
data['frac_change_colonies'] = ((data['colonies_post'] - data['colonies_pre'])
/ data['colonies_pre'])
data['frac_change_colonies'] = pm.floatX(data['frac_change_colonies'])
del data['screen protector']
# Change dtypes to int32 for GPU usage.
def change_dtype(data, dtype='int32'):
return data.astype(dtype)
cols_to_change_ints = ['sample_id', 'colonies_pre', 'colonies_post',
'morphologies_pre', 'morphologies_post', 'phone ID']
cols_to_change_floats = ['year', 'month', 'day', 'perc_reduction morph',
'phone ID', 'no case',]
for col in cols_to_change_ints:
data[col] = change_dtype(data[col], dtype='int32')
for col in cols_to_change_floats:
data[col] = change_dtype(data[col], dtype='float32')
data.dtypes
# # filter the data such that we have only PhoneSoap (PS-300) and Ethanol (ET)
# data_filtered = data[(data['treatment'] == 'PS-300') | (data['treatment'] == 'QA')]
# data_filtered = data_filtered[data_filtered['site'] == 'phone']
# data_filtered.sample(10)
data¶
def plot_colonies_data():
fig = plt.figure(figsize=(10,5))
ax1 = fig.add_subplot(2,1,1)
sns.swarmplot(x='treatment', y='colonies_pre', data=data, ax=ax1)
ax1.set_title('pre-treatment')
ax1.set_xlabel('')
ax1.set_ylabel('colonies')
ax2 = fig.add_subplot(2,1,2)
sns.swarmplot(x='treatment', y='colonies_post', data=data, ax=ax2)
ax2.set_title('post-treatment')
ax2.set_ylabel('colonies')
ax2.set_ylim(ax1.get_ylim())
plt.tight_layout()
return fig
fig = plot_colonies_data()
plt.show()

priors¶
Counts are Poisson distributed. Thus...
- Count likelihoods are Poisson distributed: $ data_{i}^{j} \sim Poisson(\mu_{i}^{j}), j \in [pre, post], i \in [1, 2, 3...] $
- Priors for Poisson parameters are discrete uniform distributed: $ \mu_{i}^{j} \sim DiscreteUniform(0, 10^{4}), j \in [pre, post], i \in [1, 2, 3...] $
- Sterilization efficacy is measured by percentage change, defined as: $ \frac{mu_{pre} - mu_{post}}{mu_{pre}} $

code¶
with pm.Model() as poisson_estimation:
mu_pre = pm.DiscreteUniform('pre_mus', lower=0, upper=10000,
shape=len(treatment_order))
pre_mus = mu_pre[data['treatment'].values] # fancy indexing!!
pre_counts = pm.Poisson('pre_counts', mu=pre_mus,
observed=pm.floatX(data['colonies_pre']))
mu_post = pm.DiscreteUniform('post_mus', lower=0, upper=10000,
shape=len(treatment_order))
post_mus = mu_post[data['treatment'].values] # fancy indexing!!
post_counts = pm.Poisson('post_counts', mu=post_mus,
observed=pm.floatX(data['colonies_post']))
perc_change = pm.Deterministic('perc_change',
100 * (mu_pre - mu_post) / mu_pre)
MCMC Inference Button (TM)¶
with poisson_estimation:
poisson_trace = pm.sample(200000)
pm.traceplot(poisson_trace[50000:], varnames=['pre_mus', 'post_mus'])
plt.show()

results¶
pm.forestplot(poisson_trace[50000:], varnames=['perc_change'],
ylabels=treatment_order, xrange=[0, 110])
plt.xlabel('Percentage Reduction')
ax = plt.gca()
ax = adjust_forestplot_for_slides(ax)

problem type 3: complicated stuff¶
example: bayesian neural networks¶
a.k.a. bayesian deep learning
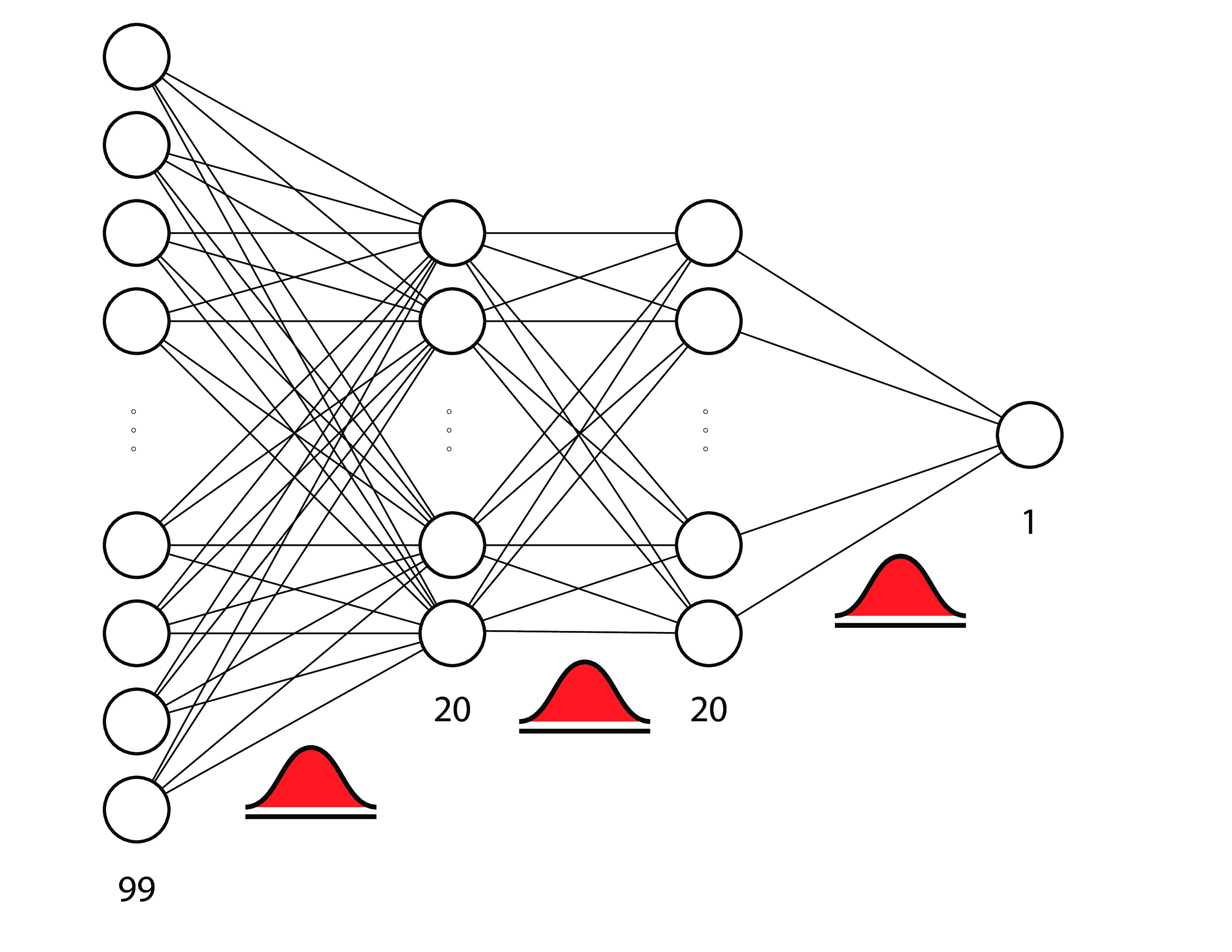
concepts featured¶
- Parameter Estimation:
- Coin Toss: Priors & Posteriors
- IC50: Link functions & Deterministic computations
- Control vs. Treatment:
- Drug IQ: One treatment vs. one control
- Phone Sterilization: Multiple treatments vs. multiple controls.
- Bayesian Neural Nets:
- Forest Cover: Parameter priors & Approximate inference.
pattern¶
- parameterize your problem using statistical distributions
- justify your model structure
- write model in PyMC3, hit the Inference ButtonTM
- interpret based on posterior distributions
- (optional) with new information, modify model structure.
bayesian estimation¶
- write a descriptive model for how the data were generated.
- original bayes: do this before seeing your data.
- empirical bayes: do this after seeing your data.
- estimate posterior distributions of model parameters of interest.
- deterministically compute posterior distributions of derived parameters.
resources¶
- John K. Kruschke's books, paper, and video.
- Statistical Re-thinking book
- Jake Vanderplas' blog post on the differences between Frequentism and Bayesianism.
- PyMC3 examples & documentation
- Andrew Gelman's blog
- Recommendations for prior distributions wiki
- Cam Davidson-Pilon's Bayesian Methods for Hackers
- My repository of Bayesian data analysis recipes.
GO BAYES!¶
- Full notebook with bonus resources: https://github.com/ericmjl/bayesian-stats-talk
- Twitter: @ericmjl
- Website: ericmjl.com